Autorisations des OGM en Europe : une législation inadaptée
Par Eric MEUNIER, Pauline VERRIERE
Publié le 13/02/2012



La législation européenne pour les procédures d’autorisation des plantes génétiquement modifiées (PGM) vient d’être évaluée par deux cabinets de consultants. Leurs rapports ont été rendus publics en octobre 2011 par la Commission européenne. L’occasion pour Inf’OGM de faire le point, à la fois sur les deux procédures d’autorisation commerciale dans l’Union européenne ; et sur certaines propositions des acteurs du dossier (entreprises, États membres, organismes européens, associations…), pour « améliorer » ce fonctionnement.
A l’heure actuelle, l’UE a dans ses dossiers 78 demandes d’autorisation commerciale de différentes PGM (cf. encadré ci-dessous). Toutes sont concernées par l’une ou l’autre des deux procédures européennes qui permettent d’obtenir une autorisation : la directive 2001/18 qui encadre la dissémination des OGM dans l’environnement ou le règlement 1829/2003 qui traite des OGM à destination de l’alimentation humaine et animale. Chacun de ces textes propose une procédure particulière d’autorisation des OGM [1].
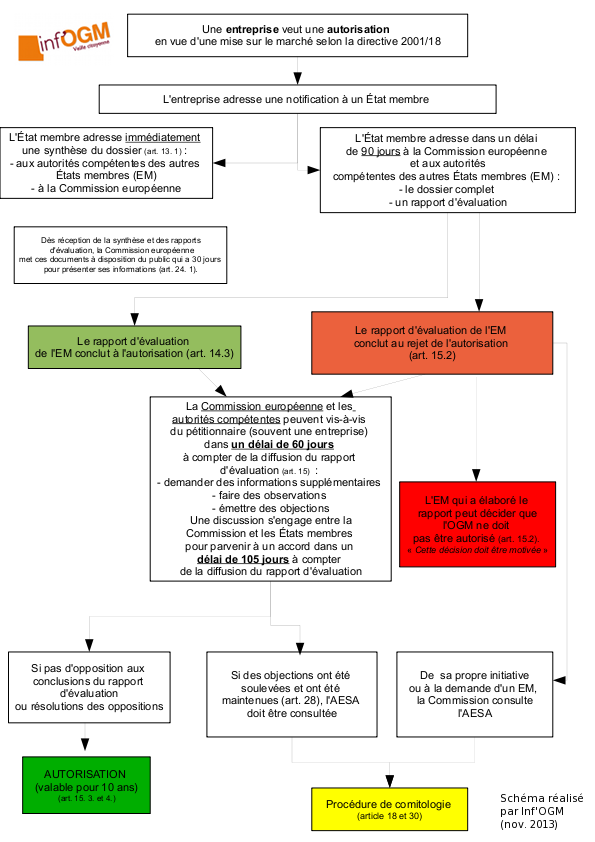
La directive 2001/18 : pour les essais et pour la mise sur le marché
La directive distingue d’un côté les essais en champs et de l’autre la mise sur le marché. Pour cette dernière, une entreprise adresse une demande (appelée notification) à un État membre (EM). Ce dernier, après avoir évalué le dossier, le transmet aux autres EM et à la Commission européenne accompagné du rapport d’évaluation [2]. Dans ce rapport d’évaluation, l’État qui a reçu la notification indique s’il va autoriser ou non la PGM. En cas de refus, l’entreprise peut déposer une seconde fois sa demande auprès d’un État membre différent. Aucune limite de nombre de dépôts n’est indiquée, permettant éventuellement à l’entreprise de déposer sa demande dans autant d’EM que nécessaire pour obtenir un rapport favorable. Une fois ce rapport favorable établi, la Commission européenne (CE) et les EM sont alors associés au processus décisionnel : s’ils sont d’accord, l’autorisation est donnée (une telle unanimité n’est jamais arrivée). S’ils ne sont pas d’accord, la demande passe alors aux mains de la CE par la procédure de comitologie (qui a récemment évolué [3]). C’est donc la CE qui va faire une proposition d’autoriser ou non l’OGM, sur la base de l’avis des experts européens réunis au sein de l’Autorité Européenne de Sécurité des Aliments (AESA). Les avis de ces derniers ont toujours permis à la CE de proposer une autorisation aux EM. La raison est que les entreprises retirent, avant avis final, les dossiers pour lesquels les experts européens vont publier un avis défavorable. En effet, du fait des discussions entre experts et entreprises sur les désaccords scientifiques, ces dernières peuvent évaluer la probabilité pour que leur demande d’autorisation reçoive un avis défavorable de la part des experts européens. A noter également que la CE a toujours suivi ces avis même dans le cas des maïs Bt11 et TC1507 pour lesquels, en 2007 et parce que poussée par le commissaire en charge des OGM à l’époque, Stavros Dimas, elle avait demandé à l’AESA un second avis. Selon les nouvelles règles de comitologie, la proposition d’autorisation ou de refus est alors soumise au vote du Comité réglementaire de la directive 2001/18, composé de représentants des États membres (des fonctionnaires donc non élus). Si ce dernier parvient à un accord à la majorité qualifiée, la décision est prise en ce sens (autorisation ou non), sinon c’est à la Commission européenne que revient la décision finale sur sa propre proposition, mais avec la possibilité de ne pas la suivre. Actuellement, des dossiers sont en cours de traitement par la procédure 2001/18, à l’image de deux demandes d’autorisations commerciales pour des œillets à couleur modifiée et tolérant des herbicides (dossier NL/2009/01 et NL/2009/02).
PGM dans l’UE : 78 demandes d’autorisation commerciale en attente
Parmi les demandes d’autorisation en attente, 26 concernent notamment la mise en culture, 64 portent sur l’alimentation humaine et/ou animale et 68 sur l’importation. Les plus anciennes datent de 1996 (coton MON531, maïs Bt11, colza Falcon GS40/90…) et la plus récente est la demande concernant la pomme de terre PH05-026-0048 dite Fortuna (cf. Les entreprises à l’offensive sur le dossier OGM). Enfin, il faut rappeler que trois PGM – le maïs Mon810, le maïs T25 et la pomme de terre Amflora – sont déjà autorisées à la culture et 31 le sont à l’alimentation humaine et/ou animale (dont 23 – soit 72%, sont du maïs).
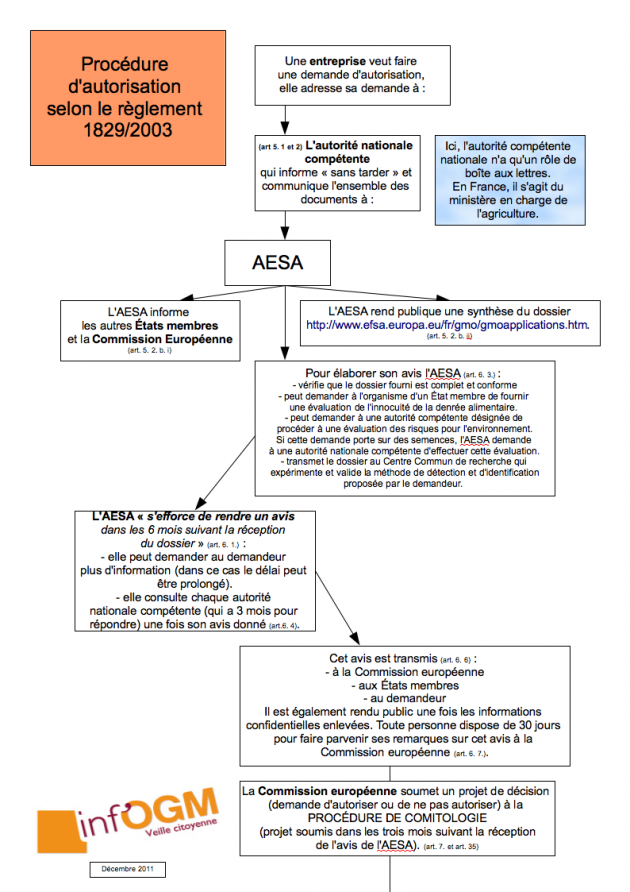
Le règlement 1829/2003 : seulement pour les demandes commerciales
Le règlement ne s’intéresse qu’aux demandes d’autorisation commerciale des OGM à destination de l’alimentation humaine et animale. A cette fin, l’entreprise adresse une demande à un État membre qui ici ne joue que le rôle de boîte aux lettres. En effet, sans regarder le dossier, l’EM le transmet à l’AESA qui informe les autres EM et la CE. L’AESA va ensuite émettre un avis scientifique concernant cette PGM. Pour élaborer cet avis, elle vérifie d’abord que le dossier est complet et conforme. Elle peut demander à un État membre de fournir une évaluation sur l’innocuité de la denrée alimentaire ou sur les risques pour l’environnement. Mais une telle consultation n’est obligatoire que si la demande porte sur des semences. Enfin, l’AESA vérifie les données, fournies par le demandeur, qui démontrent que les « caractéristiques » du produit à autoriser « ne diffèrent pas » de celles du produit conventionnel. Une fois adopté, l’avis de l’AESA est transmis à la Commission, aux EM et au demandeur [4]. Si à ce stade, chaque État membre doit être consulté, nombreux sont ceux qui regrettent que cette consultation n’interviennent qu’une fois l’avis publié [5].
Le règlement associe donc beaucoup moins les EM au processus d’autorisation d’un OGM que ne le fait la directive 2001/18. A bon droit, les EM se demandent d’ailleurs ce que deviennent leurs commentaires, et doutent de l’intérêt d’être consultés sur le contenu de l’avis scientifique si leurs observations n’ont aucune influence. En pratique, c’est donc une simple information qui est faite quand le texte prévoit bien une consultation. La suite de la procédure suit la comitologie : la Commission européenne, sur base de l’avis de l’AESA, fait une proposition d’autoriser ou non l’OGM, la soumet au Comité Permanent de la Chaîne Alimentaire et de la Santé Animale (CP CASA) qui, comme le Comité réglementaire de la directive 2001/18, est composé de représentants des États membres (là encore des fonctionnaires, et non des élus) [6]. A ce jour, seulement 11 dossiers (sur les 78 en cours) ont suivi la directive 2001/18. La quasi totalité des demandes d’autorisation est maintenant déposée selon la procédure 1829/2003, et depuis 2008, même celles concernant uniquement la culture, comme nous l’avions déjà indiqué [7].
Des législations soumises à critiques
En juin 2009, la Commission européenne a commandé une évaluation du cadre juridique européen sur les OGM. Cette évaluation, rendue public en octobre 2011, a consisté en deux études [8]. La première, coordonnée par le cabinet privé GHK Consulting, s’est intéressée aux textes encadrant la culture des PGM, à savoir la directive 2001/18 et le règlement 1829/2003. La seconde, coordonnée par le cabinet privé Agra CEAS Consulting, s’est intéressée aux textes encadrant l’alimentation, le règlement 1829/2003 et 1830/2003.
Pour l’évaluation sur les textes encadrant la culture, l’ensemble des acteurs entendus [9] considère que ces deux législations (la directive 2001/18 et le règlement 1829/2003) poursuivent les mêmes buts de protection de la santé humaine et animale, protection de l’environnement et de l’intérêt des consommateurs, tout en assurant le fonctionnement du marché. Néanmoins, tous les acteurs ne donnent pas le même poids à chacun de ces objectifs. Le rapport souligne toute la difficulté de conserver une législation cohérente face à des techniques en constante évolution. Sur les procédures d’autorisations en elles-mêmes, outre le défaut de consultation soulevé par certains États membres comme vu plus haut, le rapport met en lumière le besoin d’une meilleure préparation des dossiers en amont de leur dépôt, préparation nécessitant des discussions entre l’AESA et les autorités compétentes et les demandeurs afin que leurs dossiers soient immédiatement conformes aux exigences de l’UE. Une meilleure communication entre les différents acteurs permettrait ainsi d’éviter une demande de compléments concernant le dossier et donc le temps passé à demander, recevoir et évaluer ces compléments. Les États membres ont exprimé leur regret de n’avoir que trop peu de temps (trois mois) pour être consultés sur des dossiers plus que volumineux, qui s’accumulent et qu’ils jugent pourtant bien souvent incomplets.
En conclusion, selon le rapport, les deux textes ne sont pas adaptés à la situation. « Certains États membres » souhaitent que des considérations socio-économiques et éthiques soient incluses dans les évaluations, de même que des considérations sur les caractéristiques agro-écologiques régionales. Autre point, demander une autorisation à la culture selon le 1829/2003 implique deux choses : moins de dialogue entre AESA et États membres et des décisions nationales d’interdiction (mesure d’urgence) plus difficiles à mettre en œuvre que celles de la directive 2001/18 (clause de sauvegarde). Certes, la directive et le règlement avaient initialement un champ de compétence distinct, mais l’utilisation du principe « one key, one door » (une clé, une porte), officiellement pour des raisons de simplification des procédures administratives, permet aux demandeurs d’utiliser indifféremment l’une ou l’autre procédure (sous condition que leur demande concerne des destinations – culture, alimentation humaine ou animale – couverte par les textes législatifs). Les demandeurs ne se sont donc pas trompés sur toutes les conséquences de cette « facilitation » administrative [10]. Dernier point d’importance, le rapport souligne le cas des nouvelles techniques de biotechnologies pour lesquelles des discussions sont en cours afin d’établir si les textes existants s’appliquent à ces techniques ou non [11]. Un chantier supplémentaire qui va générer de nombreuses discussions à n’en pas douter !
Selon le rapport, le coût de l’ensemble de ces procédures d’autorisation pour une entreprise est de 6,8 millions d’euros par dossier déposé [12]. Une partie de ce coût étant liée à l’évaluation, les auteurs considèrent qu’un renforcement financier de l’AESA permettrait à cette dernière d’être plus efficace en disposant, a priori, de plus de moyens et permettrait donc, pour les entreprises, d’obtenir des autorisations plus « robustes ». Différentes solutions sont envisagées pour ce renforcement financier de l’AESA. Première hypothèse : facturer l’évaluation aux demandeurs d’autorisation. Selon les auteurs du rapport, le coût global pour arriver à la commercialisation d’une nouvelle caractéristique par génie génétique se situe entre 200 et 400 millions d’euros. Un surcoût de 1,5 à 3% ne saurait donc être dissuasif. Seconde hypothèse : facturer aux États membres le « travail » supplémentaire donné à l’AESA, par exemple lorsqu’un État membre décide de prendre une clause de sauvegarde ou une mesure d’urgence. A noter que dans ce cas, les auteurs du rapport ne discutent pas de l’aspect dissuasif d’un tel coût… Faire supporter aux États membres (donc entre autre aux citoyens opposés aux OGM) le coût de leurs objections scientifiques à la commercialisation de PGM : voilà une proposition que les entreprises de biotechnologies n’auraient peut-être même pas osé mettre sur la table !
[1] Ces procédures sont disponibles sous forme de schéma : pour la directive 2001/18 : http://www.infogm.org/spip.php?arti… et
http://www.infogm.org/spip.php?arti… et pour le règlement 1829/2003 : http://www.infogm.org/spip.php?arti… et [http://www.infogm.org/spip.php?arti…
[2] La Commission européenne, à ce stade, doit transmettre le rapport d’évaluation et la synthèse du dossier au public qui a trente jours pour faire parvenir ses remarques sur la demande.
[3] Verrière, P., « Nouvelle comitologie à l’UE : on cherche le « plus » démocratique », Inf’OGM n°109, mars / avril 2011
[4] L’avis, dans lequel toute information jugée confidentielle aura été enlevée, sera ensuite transmis au public, lequel aura 30 jours pour faire remonter ses remarques à la Commission européenne.
[5] Positions exprimées dans le rapport « Évaluation du cadre juridique européen concernant la culture des OGM sous la directive 2001/18 et le règlement 1829/2003 », p 24. Rapport intégral seulement disponible en anglais sur : http://ec.europa.eu/food/food/biote…, résumé français sur : [http://ec.europa.eu/food/food/biotechnology/evaluation/docs/gmo_cultivation_executive_summary_fr.pdf]
[6] Dans l’ancienne procédure de comitologie, si ce groupe ne parvenait pas à un accord, le vote était alors soumis au Conseil des ministres de l’Union européenne (ministres européens de l’agriculture ou de l’environnement), issu donc d’un processus démocratique. Ce volet démocratique a été épuré de la nouvelle procédure de comitologie.
[9] Le rapport intitulé « Évaluation du cadre juridique européen concernant la culture des OGM sous la directive 2001/18 et le règlement 1829/2003 » est issu de la consultation de différentes parties prenantes à la question des OGM : AESA, États membres, ONG environnementalistes, entreprises notifiantes, associations d’industriels…
[12]

« Vaccin » à ARN messager auto-amplificateur : après l’Homme, le Canard - 4 mars 2025

Algues OGM : une matière première en devenir pour l’industrie - 4 février 2025

La proposition polonaise sur les brevets et les OGM plonge les États membres dans le doute - 28 janvier 2025
En Australie, Oxitec et le CSIRO développent des moustiques OGM - 21 janvier 2025
Faq
Quels sont les OGM autorisés (culture et importation) dans l’UE ? - 15 octobre 2018

Quels sont les OGM en cours d’autorisation (culture et importation) dans l’UE ? - 10 avril 2015

Le cadre règlementaire européen des OGM - 17 octobre 2014

Autorisation et procédures d’autorisations des OGM dans l’UE - 17 octobre 2014
A lire également
GMO Canola: The Canadian government comes to the rescue of Cibus - 13 novembre 2020
Etat des lieux des OGM en Europe - 3 octobre 2019
Procédure d’évaluation et d’autorisation des OGM selon la directive 2001/18 - 6 novembre 2013