Actualités
Modifier génétiquement une plante est loin d’être anodin (suite)
Par Eric MEUNIER
Publié le 01/06/2017





Une nouvelle publication (juin 2017) fait état de centaines de mutations inattendues chez des souris modifiées par Crispr-Cas9. Ce n’est qu’une confirmation, pour Inf’OGM, de la série d’articles écrits à l’été 2016, dont nous republions ici une version actualisée.
Dans un précédent article sur ce sujet (1° volet), Inf’OGM a abordé la question des mutations et épimutations générées à chacune des étapes préalables à quelque technique de modification génétique que ce soit. Nous allons ici aborder la question des effets non intentionnels ou limites générés par les étapes postérieures : sélection des cellules ayant été réellement modifiées, régénération d’une plante entière à partir de ces cellules modifiées et croisement de cette plante génétiquement modifiée avec une variété « élite » (étape couramment appelée « rétrocroisement »).
Modifier le génome d’une plante signifie travailler in vitro sur des cellules végétales qui devront avoir été préparées et mises en culture, puis avoir reçu le matériel capable de générer la (ou les) modification(s) génétique(s) désirée(s).
Sélectionner et régénérer les cellules « modifiées » n’est pas sans effet
Du fait de leur faible, voire très faible, efficacité de transformation [1], les techniques de modification génétique impliquent en aval de sélectionner les cellules ayant effectivement été modifiées. Cette sélection se fait en utilisant des marqueurs qui permettent de différencier les cellules : gène de résistance à des antibiotiques ou à des herbicides (les cellules sont alors plongées dans un bain d’antibiotiques ou d’herbicides et seules les cellules réellement modifiées survivent), gène qui permet la croissance de la cellule en présence d’une molécule habituellement toxique ou encore gène qui permet aux cellules d’utiliser une source de carbone normalement non métabolisable… ou tout autre marqueur qui sera persistant ou supprimé en bout de course [2]. Mais la suppression de ces marqueurs de sélection se fait par des techniques plus ou moins fiables et précises et qui donc peuvent induire potentiellement des toxicités cellulaires et autres réarrangements chromosomiques [3], laisser des empreintes [4] et produire des sites de recombinaisons (lieux d’échanges de séquence génétique entre deux brins d’ADN) aux effets non prévisibles [5]. Des techniques qui ne sont par ailleurs pas forcément possibles pour toutes les espèces végétales.
A partir de ces cellules sélectionnées, des plantes doivent ensuite être régénérées. Une nouvelle étape de cultures cellulaires qui fera appel à diverses hormones de synthèse, et pourra aussi induire à nouveau des mutations et des épimutations avec réarrangements ou non de chromosomes [6].
Qu’il s’agisse de l’étape de modification génétique en elle-même, des étapes préalables, ou des étapes postérieures, toutes induisent des mutations ou épimutations, appelées effets hors-cible. Mais, on peut entendre dans les couloirs que finalement ces (épi)mutations ne seraient pas présentes dans la plante commercialisée. La raison ? L’étape suivante, dite de rétrocroisement, permettrait de se débarrasser de tous ces effets non intentionnels…
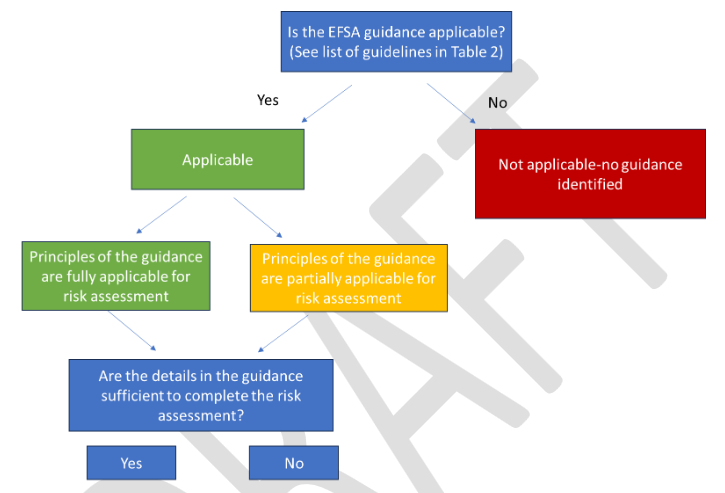
La théorie à la base du rétrocroisement
Rappelons le principe général : une entreprise qui souhaite mettre sur le marché une variété génétiquement modifiée (qu’elle soit légalement considérée comme OGM ou pas) ne va pas modifier directement le génome d’une des variétés « élites » mais celui d’une variété plus commune. Une fois la modification obtenue, l’entreprise va alors croiser une première fois la variété commune génétiquement modifiée (GM) avec la variété élite commercialement intéressante. Elle recroisera ensuite les descendants avec la variété élite initiale jusqu’à ce que ces descendants soient considérés (à partir d’une analyse statistique) quasiment similaires (on parle de variété quasi-isogénique ou de ségrégants négatifs) à la variété élite. La différence entre la variété élite et la nouvelle variété est supposée n’être « quasiment » que la seule séquence modifiée. Le GNIS explique ainsi que, au dernier croisement, « la part [du génome provenant] de la lignée élite est de 96,88%, on estime alors que la lignée obtenue est suffisamment proche de la lignée élite. On tend vers l’obtention d’une lignée isogénique, ne différant de la lignée élite que par un seul gène [celui contenant la modification] » [7]. On notera ici qu’un pourcentage de 96,88 % laisse encore beaucoup de place aux mutations, épimutations et réarrangements potentiels pour certaines plantes cultivées dont le génome est très grand comme le blé : pour environ 17 milliards de paires de base constituant le génome du blé, les 3,12% restants représentent tout de même plus de 500 millions de paires de bases…
Les limites de la théorie
La théorie sous-jacente au « nettoyage » des effets hors-cible grâce au rétrocroisement se fonde sur l’hypothèse suivante : les effets non intentionnels apparus aux étapes antérieures sont suffisamment éloignés de la région où a eu lieu la modification génétique désirée. En effet, plus ces effets hors-cibles sont proches, plus la probabilité est grande que les effets non désirés soient transmis avec la modification génétique à chaque croisement. D’un nombre minimum de sept, le nombre de croisements nécessaires pourrait alors monter à plus de 14 pour essayer de s’en débarrasser.
Mais, outre ce problème de proximité des effets hors-cible avec la séquence génétique modifiée, la confiance que l’on peut avoir dans cette théorie est également relativisée par deux autres phénomènes biologiques. D’une part, des séquences génétiques peuvent exister et évoluer en blocs plus ou moins importants. Ces blocs étant transmis tels quels à la descendance, les effets hors-cibles contenus dans ces blocs resteront alors avec la séquence génétique modifiée à chaque croisement. D’autre part, certaines séquences génétiques ont la capacité de « s’imposer » lors de la formation des gamètes. Dites « égoïstes », ces séquences vont être contenues dans un plus grand nombre de gamètes qu’attendu et, de génération en génération, elles seront toujours présentes dans la descendance. Si les effets hors-cible apparaissent dans de telles séquences, il sera moins aisé de s’en débarrasser [8].
Ces éléments impliquent donc qu’une nécessaire vérification au cas par cas paraît nécessaire. A l’heure actuelle, seule la législation sur les plantes transgéniques requiert de telles vérifications au cas par cas avec un ensemble d’analyses le plus complet qu’il soit (avec des déficiences néanmoins rappelons-le). En effet, le séquençage du génome ne permet pas de répondre à la question de la présence ou non d’effets hors-cible du fait des limites inhérentes à ce type d’analyse (voir encadré ci-dessous).
Nous avons, au cours de ces deux articles, évoqué les mutations et épimutations parmi les effets non intentionnels de ces biotechnologies modernes. On peut dès lors observer que prétendre détecter et éliminer tous les effets non intentionnels des nouvelles techniques relève plus de l’acte de foi que de la science établie. Surtout, face à ce constat des effets potentiels, des limites du rétrocroisement et de celles du séquençage, on peut douter en toute bonne foi de la capacité des sélectionneurs à éliminer tous les effets hors-cible puis à détecter efficacement et rapidement les mutations et épimutations restantes par séquençage. On peut dès lors s’étonner du silence du Comité scientifique du Haut Conseil des Biotechnologies (HCB) sur tous ces points dans son premier rapport publié en février 2016 sur les impacts liés aux nouvelles techniques de modification génétique.
Un constat renforcé par une publication récente datée de juin 2017 faisant état de centaines de mutations inattendues chez des souris modifiées par Crispr-Cas9 [9]. Des centaines de mutations passaient inaperçues car, selon les auteurs, les algorithmes informatiques utilisées se restreignent aux seuls « sites d’effets hors-cible possibles, mais ils peuvent rater des mutations ». Les auteurs estiment donc que seul le séquençage intégral du génome permettrait de détecter toutes les mutations hors-cible, une technique qui n’est pourtant pas parfaite non plus (voir encadré ci-dessous)…
Le séquençage et les outils informatiques associés ? Tout sauf un gage de certitude !
Le 7 avril, lors de l’audition organisée par l’Office parlementaire sur l’évaluation des choix scientifiques et techniques (OPECST), André Choulika, pdg de Cellectis, affirmait à propos des effets hors-cible « reséquencer intégralement [le génome des plantes], c’est vachement important […] parce que dans l’approbation, on vous redemande la séquence intégrale ». Sauf que, à bien y regarder, les résultats de séquençage obtenus sont loin d’une fiabilité absolue.
Le séquençage dit de « nouvelle génération », est aujourd’hui relativement peu cher et rapide. Mais plusieurs « problèmes » émaillent sa mise en œuvre, la lecture et l’utilisation des résultats.
Tout d’abord, plusieurs étapes du séquençage lui-même « parasitent » la fiabilité du résultat final. Il faut savoir extraire correctement l’ADN, le découper en morceaux puis séquencer ces derniers à l’aide de diverses plateformes et méthodes. Or, ces plateformes et méthodes sont assez différentes les unes des autres, tant en termes de limites que de fiabilité de résultats [10].
Il faut ensuite lire ces résultats en tentant de remettre bout à bout les séquences lues pour reconstituer le génome en entier. Puis, les séquences obtenues sont comparées avec d’autres considérées comme « de référence » et stockées dans des bases de données qui contiennent déjà elles-mêmes des erreurs [11].
Autant d’étapes qui introduisent une importante imprécision dans les résultats finaux de détection d’effets non intentionnels et donc d’évaluation des risques, les incertitudes quant aux séquences augmentant avec des génomes polyploïdes ou aux nombreuses séquences répétées [12]. Sans compter qu’en bout de course, les mutations repérées peuvent ne pas s’avérer de la même importance biologique [13]…
De nombreux articles résument les difficultés rencontrées à chaque étape, comparent les méthodes, plateformes [14] et logiciels associés [15], discutent de normes de référence (« gold standards ») et normes à mettre en place pour fiabiliser le processus complet [16]. Bref, comme le soulignent ces auteurs, autant de techniques et d’étapes qui sont en cours d’amélioration car non parvenues à maturité, en pleine évolution et nécessitant donc nombre de vérifications pour des évaluations au cas par cas.
Selon de nombreux chercheurs, savoir faire face à l’accumulation de très nombreux résultats (un de ces fameux « big data »), dont certains avec de multiples erreurs, et les utiliser avec rigueur est un des défis de la biologie moléculaire actuelle. D’ailleurs, devant le scepticisme soulevé face à tout résultat de séquençage, les demandes minimales des relecteurs d’articles scientifiques sont telles que de plus en plus de chercheurs sont maintenant obligés de présenter des résultats de séquençages de plus longues séquences pour assurer du sérieux de leurs résultats bruts [17].
[1] « Less is more : strategies to remove marker genes from transgenic plants », Yau, Y.Y. et al, (2013), BMC Biotechnology.
[2] « Alternatives to Antibiotic Resistance Marker Genes for In Vitro Selection of Genetically Modified Plants – Scientific Developments, Current Use, Operational Access and Biosafety Considerations », Breyer et al (2014) Critical Reviews in Plant Sciences, Vol 33, Issue 4, 286-330
« Suitability of non-lethal marker and marker-free systems for development of transgenic crop plants : present status and future prospects », Manimaran et al (2011) Biotechnol Adv. 29(6), 703-14
« Effects of antibiotics on suppression of Agrobacterium tumefaciens and plant regeneration from wheat embryo », Han, S-N. et al, (2004), Journal of Crop Science and Biotechnology 10, 92-98.
[3] Certains de ces systèmes peuvent persister comme éléments circulaires extra-chromosomiques durant plusieurs générations (ex. du blé).
[4] utilisables en identification (ex : empreinte d’excision de transposon, recombinaisons) de l’évènement de transformation
[5] Biotechnology 13., ibid.
[6] « Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications », Neelakandan, A.K. et al, (2012), Plant Cell Reports 31, 597-620
« Regeneration in plants and animals : dedifferentiation, transdifferentiation, or just differentiation ? », Sugimoto, K. et al, (2011), Trends in Cell Biology 21, 212-218.
[8] « Distorsions de ségrégation et amélioration génétique des plantes (synthèse bibliographique) », Diouf, F.B.H. et al , (2012), Biotechnologie Agronomie Société Et Environnement, 16(4), 499-508
« Quantitative trait locus mapping can benefit from segregation distortion », Xu, S. (2008), Genetics, 180(4), 2201-2208
« Genetic map construction and detection of genetic loci underlying segregation distortion in an intraspecific cross of Populus deltoides », Zhou, W et al, (2015), PLoS ONE, 10(5), e0126077
[9] « Unexpected mutations after CRISPR–Cas9 editing in vivo », K.A. Schaefer et al, (juin 2017), Nature Methods, 14(6), 547-548
[10] « Next generation sequencing technology : Advances and applications », Buermans, H.P.J. et al, (2014), Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, 1842(10), 1932-1941
« Next-generation sequencing platforms », Mardis, E.R. (2013), Annual Review of Analytical Chemistry, 6(1), 287-303
« Applications of next-generation sequencing. Sequencing technologies – the next generation », Metzker, M.L. (2010), Nature Reviews Genetics, 11(1), 31-46.
[11] « Next-generation sequence assembly : four stages of data processing and computational challenges », El-Metwally, S. et al, (2013), PLoS Comput Biol 9, e1003345
« Systematic comparison of variant calling pipelines using gold standard personal exome variants », Hwang, S. et al, (2015), Scientific reports 5, 17875
« Sequence assembly demystified », Nagarajan, N. et al, (2013), Nat Rev Genet 14, 157-167.« Improving the quality of genome, protein sequence, and taxonomy databases : a prerequisite for microbiome meta-omics 2.0 », Pible, O. et al, (2015). Proteomics 15, 3418-3423
« Theoretical analysis of mutation hotspots and their DNA sequence context specificity », Rogozin, I.B. et al, (2003), Mutation Research/Reviews in Mutation Research 544, 65-85
[12] « Sequencing technologies and tools for short tandem repeat variation detection », Cao, M.D. et al, (2014), Briefings in Bioinformatics
[13] « Open chromatin reveals the functional maize genome », Rodgers-Melnick, E. et al, (2016). Proceedings of the National Academy of Sciences 113, E3177-E3184
« Evolutionary patterns of genic DNA methylation vary across land plants », Takuno, S. et al, (2016), Nature Plants 2, 15222.
[14] « Systematic comparison of variant calling pipelines using gold standard personal exome variants », Hwang, S., et al, (2015), Scientific reports 5, 17875
« Principles and challenges of genome-wide DNA methylation analysis », Laird, P.W. (2010), Nature Reviews Genetics 11, 191-203
« Performance comparison of whole-genome sequencing platforms », Lam, H.Y.K. et al (2012), Nat Biotech 30, 78-82
« Low concordance of multiple variant-calling pipelines : practical implications for exome and genome sequencing », O’Rawe, J. et al, (2013), Genome Medicine 5, 1-18.
[15] « Next-generation sequence assembly : four stages of data processing and computational challenges », El-Metwally, S. et al, (2013), PLoS Comput Biol 9, e1003345
[16] « Rapid evaluation and quality control of next generation sequencing data with FaQCs », Lo, C.-C. Et al, (2014), BMC Bioinformatics 15, 1-8
[17] « Droplet barcoding for massively parallel single-molecule deep sequencing », Lan, F. et al, (2016), Nat Commun 7